Learning Objectives
- Identify the biochemical shunts that occur when the PDH complex is inactive.
- Recognize the clinical triad of lactic acidosis, neurologic defects, and elevated alanine.
- Explain the rationale behind using ketogenic nutrients in management.
1. Biochemical Pathophysiology
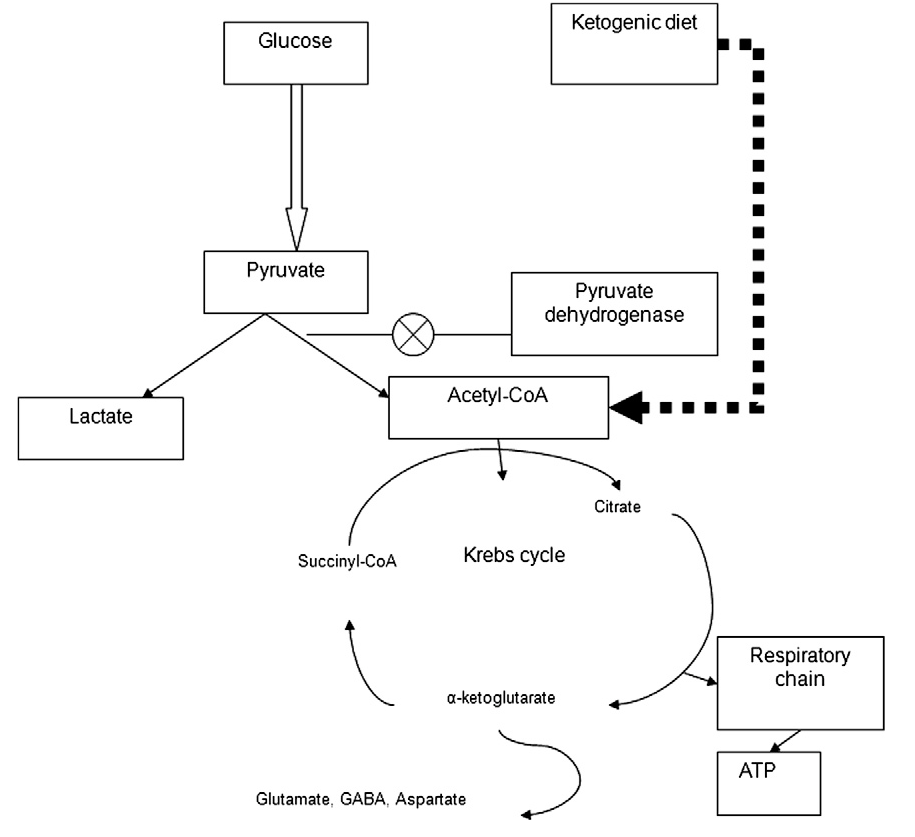
When the PDH complex is deficient, pyruvate cannot be converted to Acetyl-CoA. This creates a metabolic “logjam,” forcing pyruvate into alternative pathways to regenerate NAD+ or dispose of nitrogen.
- Lactate Shunt: Pyruvate is converted to lactate by Lactate Dehydrogenase (LDH), leading to Lactic Acidosis.
- Alanine Shunt: Pyruvate is transaminated to alanine by Alanine Aminotransferase (ALT), resulting in Hyperalaninemia.
2. Clinical Presentation
As an X-linked disorder, it typically presents in infancy. Because the brain relies heavily on aerobic glucose metabolism, the lack of TCA cycle entry causes severe energy deficits in the CNS.
- Neurologic defects: Poor feeding, developmental delay, seizures, and structural brain abnormalities.
- Metabolic: Chronic lactic acidosis (low pH, high anion gap).
Biochemical Correlation: Purely Ketogenic Amino Acids
In PDH deficiency, we must bypass the PDH step by providing substrates that enter the TCA cycle directly as Acetyl-CoA or Acetoacetate. Lysine and Leucine are the only two purely ketogenic amino acids; their metabolism does not produce pyruvate, thus avoiding further lactic acid production.
3. Treatment Strategy
Management focuses on reducing the pyruvate load and maximizing the efficiency of any remaining enzyme activity.
- Dietary: High-fat, low-carbohydrate diet (Ketogenic diet).
- Amino Acids: Supplementation with Lysine and Leucine.
- Cofactors: High-dose Thiamine (B1) and Lipoic Acid to optimize residual E1 and E2 subunit function.
Activity
Activity
You must be logged in to post a comment.